Research Article
A Structure-Driven Statistical Framework for Rare Variant Association Analysis: Unifying Burden Tests and SKAT with a Decision Pipeline for Method Selection 
 Author
Author  Correspondence author
Correspondence author
Computational Molecular Biology, 2026, Vol. 16, No. 4
Received: 16 May, 2026 Accepted: 20 Jun., 2026 Published: 05 Jul., 2026
With the advent of high-throughput sequencing technologies, research in complex trait genetics is expanding from the GWAS paradigm focused primarily on common variants to full-spectrum analyses that also encompass rare variants. Compared with single-marker tests, rare variant analysis faces substantial challenges in statistical power and model specification because of low allele frequencies, strong effect heterogeneity, and contamination by non-causal variants. To address these issues, set-based aggregation tests have gradually become the dominant analytical strategy, with burden tests and the Sequence Kernel Association Test (SKAT) constituting two core methodological tracks. In this study, we develop a unified statistical framework to systematically compare burden tests and the SKAT family of methods in terms of their modeling assumptions, conditions of applicability, and performance boundaries. We show that the differences between these two classes of methods fundamentally arise from different characterizations of effect structure within a gene region: burden tests, through linear aggregation, are more suitable for scenarios dominated by concordant effect directions, whereas SKAT, through variance-component modeling, is better suited to heterogeneous effect directions and sparse causal architectures. On this basis, we further propose a decision pipeline for method selection based on genetic architecture, reframing method selection as a problem of causal architecture identification and thereby enabling a shift from heuristic choice to model-driven decision-making. Through simulation and empirical analyses, we show that different methods have clearly defined optimal regions within the space of effect-direction concordance and causal proportion, whereas SKAT-O and multi-kernel methods exhibit strong robustness when the underlying structure is unknown. Drawing on case studies from both human and crop genetics, this study further demonstrates the potential of rare variant analysis for elucidating functional mechanisms and supporting applied research. Overall, this study integrates rare variant association analysis into a structure-oriented statistical inference framework, providing a unified and operational theoretical foundation for method selection, result interpretation, and cross-study application in complex trait research.
Understanding the genetic basis of complex traits has undergone a paradigm evolution in statistical genetics, from linkage analysis and candidate-gene strategies to modern GWAS, and is now further expanding from the classical “common variant-small effect” GWAS framework toward a continuous genetic spectrum encompassing “rare variants-moderate to large effects” (Fang and Wu, 2026). With the widespread adoption of whole-exome and whole-genome sequencing (WES/WGS), researchers are now able to interrogate both common and rare variants across the genome, thereby advancing the study of genetic architecture in complex traits to a more refined stage (Lee et al., 2012a; Rajabli and Kunkle, 2023). Although GWAS has successfully identified numerous trait-associated loci, it primarily focuses on common variants and still leaves a substantial gap in explaining the heritability of complex traits, namely the so-called “missing heritability” problem (Falk et al., 2023; Rajabli and Kunkle, 2023). In this context, rare variants, which are often constrained by purifying selection, tend to exhibit larger allelic effects, stronger allelic heterogeneity, and more pronounced functional enrichment, making them an important complementary source for linking statistical association to biological mechanisms (Ionita-Laza et al., 2013).
However, the statistical analysis of rare variants faces fundamental challenges. Within a single-variant framework, statistical power scales approximately with the joint contribution of sample size n, allele frequency p , and effect size β (Power∝n⋅p(1-p)β2). When p is extremely low, even large-scale samples may fail to provide sufficient statistical signal (Lee et al., 2012a). In addition, complex traits commonly exhibit effect heterogeneity and noise introduced by non-causal variants, which limit the power of single-variant tests and may also introduce type I error miscalibration (Hecker et al., 2020). Therefore, the conventional GWAS framework cannot be directly extended to rare variant settings.
To overcome these limitations, set-based aggregation analysis has become the dominant strategy. This approach aggregates multiple rare variants within a gene or functional region into a single statistical unit, thereby effectively increasing the effective sample size from a statistical perspective while aligning the analysis with biologically meaningful functional units (Lee et al., 2012b; Ionita-Laza et al., 2013). In statistical modeling, these methods typically incorporate weighting schemes that integrate allele frequency and functional annotation, and combine them with linear or generalized linear mixed models to control for population structure, relatedness, and sample imbalance, thereby enabling more robust inference in real-world data (Zhang et al., 2019; Hecker et al., 2020).
From a unified statistical genetics perspective, rare variant analysis can be understood as an inferential problem concerning set-level genetic effects, namely the modeling of the overall effect structure of a group of variants. This complements GWAS (association estimand), heritability estimation (variance estimand), and fine-mapping (causal probability estimand), jointly forming a multi-layered inference system for complex trait genetics. Under this framework, the core distinction among aggregation tests is no longer simply a matter of method choice, but rather reflects different forms of effect representation under different statistical assumptions.
Specifically, existing methods can be broadly divided into two statistical tracks. Burden tests assume that variant effects within a set are directionally consistent and collapse them into a single weighted effect, corresponding to the estimation of an average effect under a fixed-effect model. In contrast, the Sequence Kernel Association Test (SKAT) treats site-specific effects as random variables and characterizes their overall variability through a variance-component model, thereby naturally accommodating heterogeneous effect directions and sparse causal architectures (Lee et al., 2012a). On this basis, methods such as SKAT-O achieve robust approximation to unknown genetic architectures by adaptively weighting fixed-effect and random-effect models (Lee et al., 2012b).
Although this methodological framework is relatively mature, its performance boundaries under different genetic architectures and study designs still lack systematic characterization. In particular, under the joint influence of key factors such as causal proportion, effect-direction concordance, weight misspecification, sample size, case-control imbalance, and multi-ancestry data integration, the optimal regimes of application for different methods remain unclear (Zhang et al., 2019; Falk et al., 2023). Therefore, a systematic comparison under a unified framework is needed to clarify these issues and define their practical decision pathway.
Accordingly, this study is organized around the triadic objective of power, robustness, and interpretability. First, through theoretical derivation and simulation analysis, we systematically compare the statistical performance of burden tests and the SKAT family under different genetic architectures and data conditions. Second, through empirical analyses of human and crop genetic data, we evaluate the effects of population structure, batch effects, and functional annotation quality on inference, and further examine calibration strategies under extreme imbalance and multi-ancestry settings (e.g., saddlepoint approximation and Firth correction). Finally, within a unified statistical framework, we propose a reusable practical decision system that incorporates variant selection, weighting design, method choice, and reporting standards, thereby providing systematic methodological guidance for complex trait genetics research. Within this framework, rare variant analysis should no longer be viewed as a supplementary technique to GWAS, but rather as an independent layer of inference within the causal inference chain of complex traits. Together with single-variant analysis and fine-mapping, it contributes to the progressive convergence from association signals to mechanistic interpretation.
1 Statistical Challenges in Rare Variant Association: From Single-Variant Power Limits to Set-Level Inference
1.1 Power limits and information bottlenecks in single-variant analysis
Within the classical single-variant association framework, statistical power is fundamentally constrained by both allele frequency and sample size. Under an additive model, the genotype variance at a locus is given by Var(G)=2p(1-p), and the non-centrality parameter (NCP) of the test statistic in a generalized linear model can be approximated as:
λ∝neff⋅2p(1-p)β2,
For binary traits, this expression is further multiplied by the case proportion factor π(1-π) (Lee et al., 2014). When the allele frequency is extremely low (p≪1), the number of carriers—quantified by the minor allele count (MAC)—rapidly decreases, causing λ to decay accordingly. As a result, even very large cohorts may fail to achieve adequate statistical power (Bigdeli et al., 2014; Lee et al., 2014). This phenomenon reveals a fundamental limitation: single-variant tests exhibit “information non-scalability” in the rare variant setting.
In addition, under extreme case–control imbalance, traditional tests based on asymptotic distributions (e.g., Wald or score statistics) may become inaccurate, resulting in inflated type I error rates. Although approaches such as saddlepoint approximation and Firth bias correction can improve significance calibration, they only correct the distributional form and do not increase the effective information content (Lee et al., 2014; Wang, 2014).
In real-world data, multiple factors further weaken statistical power, including underestimation of MAC due to sequencing errors, variance inflation caused by population structure and relatedness, and systematic biases introduced by batch effects and deviations from Hardy–Weinberg equilibrium. At the genome-wide scale, stringent multiple-testing thresholds (e.g., α≈5×10-8) further compress the space of detectable signals (Bigdeli et al., 2014). More importantly, complex traits typically exhibit substantial allelic heterogeneity, meaning that different variants within the same gene may differ in both effect size and direction. This makes the single-variant framework inadequate for integrating the overall effect structure and limits its utility for mechanistic interpretation (Boutry et al., 2023a; Rajabli and Kunkle, 2023).
From a statistical inference perspective, the core question therefore shifts from detecting the effect of an individual variant to modeling and inferring the overall effect structure of a set of rare variants.
1.2 Aggregation tests: from single-variant inference to the set-level estimand
To overcome the limitations of single-variant analysis, rare variant association research has introduced set-level aggregation tests, namely rare variant aggregation tests (RVAT). This strategy takes a gene or functional region as the unit and integrates information from multiple variants into a single statistical object, thereby increasing the effective signal strength statistically while remaining biologically consistent with functional units (Lee et al., 2014; Rajabli and Kunkle, 2023).
A typical weighted burden statistic can be written as
.png)
where the weights wj are typically defined on the basis of allele frequency and functional annotation (e.g., Beta weights, CADD scores, or prioritization of LoF variants) to prioritize potentially functional variants (Boutry et al., 2023a). In a regression framework, testing Y~Tburden+C allows direct assessment of the set-level effect.
The essence of this methodological shift lies in elevating the inferential target from a “single-variant effect” to a “set-level genetic effect.” Within a unified statistical genetics framework, this layer complements the previously discussed GWAS (association layer), heritability estimation (variance layer), and fine-mapping (causal probability layer), together constituting a multi-level inference system for complex trait genetic analysis.
1.3 Two statistical tracks: effect-structure assumptions and methodological divergence
In set-level inference, the core of methodological divergence lies in different assumptions about variant effect structure. This can be expressed in a unified form:
Y=Xβ+ε,
where β=(β1,…,βm) is the vector of variant effects within a set. Different methods correspond to different modeling assumptions:
(1) Burden tests: fixed-effect model
Assuming that variant effects within a set are directionally consistent:
βj≈β
the aggregate effect is formed through linear summation, and its statistical power can be approximated as:
.png)
This method achieves optimal power when the causal proportion is high and the effect directions are concordant (Lee et al., 2014).
(2) SKAT: variance-component model
Assuming that variant effects follow a random distribution:
βj∼N(0,τ2)
the test statistic is constructed through the kernel matrix K=GWGT:
.png)
Its effective signal is proportional to ∑wj2βj2 and is therefore more robust to effect-direction heterogeneity and sparse causal architectures (Lee et al., 2014; Rajabli and Kunkle, 2023).
It can thus be seen that burden tests and SKAT are not simply interchangeable alternatives, but rather two statistical projections of the same causal space under different assumptions about effect structure. When the true genetic architecture is unknown, adaptive methods such as SKAT-O achieve robust performance by combining the two models in a weighted manner (Lee et al., 2012b; Pan et al., 2014).
2 Burden Tests: A Causal Signal Compression Model Based on Linear Aggregation
2.1 Core principle
Burden tests are designed to address the limited power of testing individual rare variants one by one. Their core idea is to linearly aggregate the effects of multiple rare variants within the same gene or functional region, thereby increasing the effective signal strength statistically (Lee et al., 2014; Guo et al., 2018; Ziyatdinov et al., 2024).
This method carries a key implicit assumption: that the majority of rare variants within the same set affect the phenotype in the same direction (directional consistency). For example, loss-of-function (LoF) variants or highly deleterious missense variants typically produce similar functional consequences within the same gene.
Statistically, variants within a set are collapsed into an individual-level burden score:
.png)
and are then tested in a regression model:
.png)
Here, wj denotes the weight, and S denotes the variant set (mask).
Under this framework, the non-centrality parameter of the burden test can be approximated as:
.png)
This indicates that signal accumulates at the first-order linear level: when effect directions are consistent, the signal is strengthened; when effect-direction heterogeneity or neutral variants are present, ∑wjβj may cancel out, leading to reduced power (Lee et al., 2014; Pan et al., 2014). This mechanism corresponds to the advantage of burden tests in the “high concordance–high causal proportion” region of Figure 1.
.png) Figure 1 Phase diagram of rare variant association methods under different genetic architectures Note: The x-axis represents effect heterogeneity (from low to high), and the y-axis represents the proportion of causal variants. Burden tests achieve optimal power when effect directions are consistent and the causal proportion is high. When effect-direction heterogeneity is present or causal variants are sparse, SKAT has a clear advantage. Omnibus methods such as SKAT-O and ACAT are near-optimal in most scenarios and show stronger robustness. This diagram provides an intuitive basis for method selection |
The weights wj are typically defined on the basis of allele frequency or functional annotation (e.g., Beta weights, prioritization of LoF variants, or CADD scores). The set S is defined through masking strategies (e.g., MAF ≤ 1% or functional filtering), which can reduce signal dilution caused by the inclusion of non-causal variants (Lee et al., 2014; Guo et al., 2018). Under extreme case-control imbalance, Firth correction or saddlepoint approximation can improve type I error control, but they do not alter the underlying information content (Wang, 2014).
From a unified statistical perspective, Figure 1 illustrates the regions of applicability of different methods under different signal structures, providing an intuitive basis for method selection.
2.2 Representative methods
Most existing burden tests are built on the same linear aggregation framework, and their differences are primarily reflected in two aspects: how variant sets are constructed and how weights are assigned to different variants. Around these two core components, different methods have developed implementation strategies with distinct emphases.
CAST (Cohort Allelic Sums Test) is the most basic class of burden-based methods. Its main idea is to transform whether an individual carries any rare variant into a binary indicator, namely .png) . This method is simple to implement and computationally efficient, but because it ignores information on both the number of variants and their effect sizes, its statistical power is easily diluted when many neutral or weak-effect variants are present (Lee et al., 2014).
. This method is simple to implement and computationally efficient, but because it ignores information on both the number of variants and their effect sizes, its statistical power is easily diluted when many neutral or weak-effect variants are present (Lee et al., 2014).
CMC (Combined Multivariate and Collapsing) on the basis of CAST, CMC introduces a grouping strategy, typically partitioning variants into several subsets according to allele frequency or functional annotation, and then constructing burden variables within each subset for joint testing. This method partially alleviates the impact of effect heterogeneity, but it still implicitly assumes directional consistency of variant effects within each subset and therefore performs less well when effect directions are inconsistent (Lee et al., 2014; Ziyatdinov et al., 2024).
Weighted sum statistics (e.g., WSS or the Madsen–Browning method), these methods further strengthen the contribution of rare variants through weighting functions, typically assigning higher weights to lower-frequency variants. As a result, they can substantially improve detection power in scenarios dominated by concordant effects and rare variants (Lee et al., 2014). The effectiveness of this class of methods depends on the consistency between the weighting scheme and the true underlying effects.
VT (Variable Threshold) method does not fix the allele frequency threshold in advance; instead, it repeatedly constructs burden statistics over a series of candidate thresholds and selects the optimal result, thereby improving adaptability to different frequency-distribution scenarios (Lee et al., 2014). This strategy can, to some extent, alleviate bias introduced by arbitrarily chosen thresholds, but it also introduces an additional multiple-testing burden.
In recent years, methods such as SBAT and NNLS-Joint have attempted to construct multiple burden components simultaneously under frameworks involving layered annotation and frequency stratification, and to perform an overall test through joint optimization. These methods improve robustness under complex variant structures while preserving an overall directional constraint on effects (Ziyatdinov et al., 2024).
It should be noted that a single burden test usually performs best when effect directions are consistent, but may fail substantially when effect directions are inconsistent or when the signal structure is complex. Therefore, in practical analyses, burden statistics are often combined with kernel-based methods or omnibus tests (e.g., SKAT-O and ACAT) to obtain more robust inference when the model specification is not fully clear (Liu et al., 2019).
2.3 Strengths and limitations
In rare variant analysis, when a set of variants contains a high proportion of true causal sites and their effect directions are largely consistent, burden tests usually show strong statistical performance. Their essence lies in the linear aggregation of effects across multiple loci, with the test statistic increasing with the square of the weighted total effect, which can be approximately expressed as
.png)
Under such conditions, namely when effect directions are concordant and the proportion of causal variants is high, signals can accumulate efficiently, allowing burden tests to outperform both single-variant tests and variance-component models in terms of statistical power (Lee et al., 2014; Guo et al., 2018).
From the perspective of methodological characteristics, burden tests have the advantages of simple structure and direct interpretability. Their test statistics can be viewed as measures of “gene-level cumulative effects,” and the results are easy to compare and summarize across studies. They are also convenient for integrative analyses in large-scale cohorts or multicenter studies.
However, the applicability of this class of methods depends strongly on its key assumptions. Once these assumptions deviate from the actual data structure, performance can decline rapidly. First, when variant effects are directionally inconsistent (for example, when risk and protective effects coexist), linear aggregation leads to signal cancellation, thereby substantially reducing statistical power. Second, the inclusion of non-causal variants further dilutes the true signal, causing the overall effect to be underestimated. In addition, if the weighting function or functional annotation does not match the true biological mechanism, systematic bias may also be introduced. Differences in allele frequency and linkage disequilibrium structure across populations can likewise affect the stability and transferability of burden statistics.
To address these issues, a series of practical strategies are commonly used. For example, stricter variant filtering or hierarchical masking strategies (such as retaining only loss-of-function variants) can be used to reduce sources of noise. Joint weighting schemes that combine allele frequency and functional annotation can improve sensitivity to potentially causal variants. When the effect structure is unknown or effect-direction heterogeneity is present, adaptive methods such as SKAT-O or ACAT can be introduced to improve overall robustness (Liu et al., 2019). In addition, under conditions of small sample size or extremely low allele counts, methods such as Firth correction or saddlepoint approximation can be used to improve the stability of parameter estimation and significance testing (Wang, 2014).
Overall, burden tests are highly efficient when their applicability conditions are met, but their performance is sensitive to assumptions about effect structure. Therefore, in practical analyses, they often need to be used in combination with more flexible methods in order to obtain more robust inferential results.
3 Sequence Kernel Association Test (SKAT) and Its Extensions
3.1 Core principle
Unlike burden tests, which rely on the assumption of effect-direction consistency, the Sequence Kernel Association Test (SKAT) models the effects of sites within a set as random variables and evaluates whether their overall variation deviates from zero under a variance-component framework. This formulation makes SKAT better suited to scenarios with effect-direction inconsistency, sparse causal variants, or contamination by neutral variants (Lee et al., 2012a).
Specifically, let the centered genotype matrix be defined as Z=[Gij-2pj], and assume that variant effects satisfy
.png)
where τ represents the overall effect variance. Under the null hypothesis H0:τ=0, the SKAT statistic can be written as
.png)
where W=diag(wj2). Equivalently, this statistic can also be expressed as
.png)
that is, a weighted sum of squared score statistics across sites.
This construction has several direct consequences: effects in different directions do not cancel each other out; non-causal variants have a relatively weaker influence on the test statistic; and the method can still retain some sensitivity when causal signals are sparse. In large samples, Q follows a mixture of chi-square distributions, and its significance is usually calculated using the Davies method or moment-matching approximations (Lee et al., 2012b). This framework can be naturally extended to linear or logistic regression models and can be combined with mixed-effects models to control for population structure and relatedness (Oualkacha et al., 2013).
3.2 Kernel functions and weighting design
The core of SKAT lies in characterizing genetic similarity between individuals through a kernel function, with the weighted linear kernel being the most commonly used form:
K=ZWZT.
(1) Weighting schemes
Weighting functions are used to reflect the prior importance assigned to different variants. Common strategies include Beta weights based on allele frequency (e.g., Beta(MAF;1,25)), stratified weights based on functional annotation (for example, assigning higher weights to LoF variants than to missense variants), and weighting schemes that incorporate predictive scores such as CADD or LOFTEE. The shared rationale behind these methods is the assumption that variants that are rarer or have clearer functional impacts are more likely to produce larger effects (Jiang et al., 2023).
(2) Kernel selection
With respect to kernel choice, the linear kernel is the default option and is suitable for modeling additive genetic effects. On this basis, IBS kernels or Gaussian kernels can also be used to capture nonlinear effects or haplotype structure. However, as kernel complexity increases, so do the degrees of freedom, which may in turn reduce statistical power when the signal-to-noise ratio is low (Falk et al., 2023). Therefore, in practical applications, the linear kernel is usually used as the primary choice, often accompanied by weighting optimization.
3.3 Methodological extensions
To improve adaptability under different genetic architectures, SKAT has been extended in multiple forms.
SKAT-O introduces a weighted combination of burden tests and SKAT,
.png)
and selects the optimal statistic over different values of ![]() . As a result, it approaches burden tests when effect directions are consistent, and reverts to SKAT when effects are heterogeneous, thereby exhibiting good adaptive performance (Lee et al., 2012b).
. As a result, it approaches burden tests when effect directions are consistent, and reverts to SKAT when effects are heterogeneous, thereby exhibiting good adaptive performance (Lee et al., 2012b).
MK-SKAT, by contrast, integrates multiple kernel functions or weighting schemes (for example, different frequency-based weights or functional annotations), and summarizes the results using combination tests such as Cauchy combination or minimum p strategies, thereby reducing the bias introduced by a single model specification (Falk et al., 2023).
In addition, the SKAT framework has been extended to a variety of data structures, including joint analysis of common and rare variants, extremely imbalanced case–control designs, and more complex settings such as survival data, multivariate phenotypes, or ordinal outcomes (Chen et al., 2013; Chen et al., 2014; Wu and Pankow, 2016; Jiang et al., 2023).
3.4 Strengths and limitations
When effect directions are inconsistent or causal variants are relatively sparse, the SKAT statistic can be approximately expressed as
.png)
and is therefore not affected by effect cancellation. As a result, SKAT is generally more advantageous than burden tests under such conditions and also shows good robustness to non-causal variants (Lee et al., 2014).
However, its performance also depends on data structure and model specification. When variant effects are highly concordant, signals cannot be efficiently aggregated, and its statistical power may be lower than that of burden tests. In addition, the results depend to some extent on the choice of weighting function and kernel function, and the statistic itself cannot be easily interpreted directly as a “gene-level effect size,” making its biological interpretation relatively indirect.
Based on these characteristics, SKAT or SKAT-O is usually regarded as a more robust choice when the effect structure is unknown or complex; by contrast, when clear prior information is available (for example, loss-of-function variants with concordant effect directions), burden tests may still be more efficient (Lee et al., 2014; Liu et al., 2019).
4 Variant Aggregation Strategies and Practical Design
4.1 Aggregation units: gene-level vs functional definitions
Rare variant association analysis first requires the definition of variant sets, the essence of which is to determine the basic unit of statistical testing. Different aggregation units directly influence the proportion of causal variants and the degree of effect-direction concordance within a set, thereby determining the applicability of subsequent testing methods (Boutry et al., 2023b).
Gene-level aggregation defines sets at the transcript or gene level and has the advantages of intuitive interpretation and easily reproducible results, especially for enrichment analyses of loss-of-function (LoF) or high-confidence pathogenic variants. However, due to substantial variation in gene length and the number of sites, this strategy often introduces a large number of neutral variants and may include functionally heterogeneous domains or transcripts, thereby weakening the signal and reducing the power of burden tests.
In contrast, functionally defined aggregation refines variant sets using functional annotations, such as protein-truncating variants (PTVs), damaging missense variants, splice sites, or regulatory elements (e.g., enhancers and transcription factor binding sites). This strategy can increase the causal proportion within a set and reduce effect-direction heterogeneity, but it also substantially increases the number and overlap of sets, thereby imposing a heavier multiple-testing burden.
In practice, a hierarchical strategy (gene → functional region → pathway) is commonly adopted, with initial screening performed at the gene level and subsequent functional refinement carried out within significant genes. This layered design improves signal resolution while controlling the statistical burden (Boutry et al., 2023b).
4.2 Variant selection and masking
Masking defines the set of variants included in the analysis, with the core objective of balancing causal proportion against sample information (Lee et al., 2014).
Minor allele frequency (MAF) thresholds represent the most basic filtering criterion. Common strategies include thresholds of ≤1%, ≤0.1%, or more stringent cutoffs, often implemented in parallel within the same gene. A lenient threshold may introduce many non-causal variants and dilute signal, whereas an overly stringent threshold reduces carrier counts and statistical power. Variable-threshold (VT) methods address this issue by adaptively selecting the optimal frequency threshold within each gene, often outperforming fixed-threshold strategies (Lee et al., 2014).
Functional filtering is used to further improve causal proportion. In coding regions, priority is typically given to PTVs and predicted deleterious missense variants, whereas in noncoding regions, filtering is based on evolutionary conservation, regulatory annotations, and tissue-specific evidence. For ultra-rare variants (e.g., singletons), they usually need to be used in conjunction with high-confidence functional annotation in order to avoid false positives caused by technical noise (Boutry et al., 2023b).
In addition, rigorous quality control (QC) is a necessary prerequisite, including sequencing depth, genotype quality, batch effects, and deviations from Hardy-Weinberg equilibrium. Otherwise, technical errors may be amplified during aggregation and misinterpreted as genetic signals (Lee et al., 2014).
4.3 Weighting design
Weights are used to modulate the contribution of different sites to the test statistic, and their design directly reflects assumptions about the underlying genetic architecture (Lee et al., 2014).
Frequency-based weights typically take the form of a Beta distribution:
.png)
so as to emphasize the contribution of rarer variants. This strategy implicitly assumes that rarer variants tend to have larger effects.
Functional annotation weights encode biological information into statistical weights, for example by assigning different weights according to variant type (e.g., PTV > missense > synonymous) or predictive scores (e.g., CADD, REVEL):
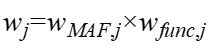
Under complex genetic architectures, a single weighting scheme may introduce a risk of mismatch. Therefore, multi-weight parallel strategies are often adopted and integrated through combination tests (e.g., Cauchy combination) or multi-kernel methods in order to improve robustness (Liu et al., 2019; Boutry et al., 2023b).
4.4 Matching design choices with statistical tests
Aggregation strategies essentially influence the power performance of different statistical tests by altering the genetic structure within a variant set, particularly the causal proportion and effect directions.
When the causal proportion within a set is high and effect directions are consistent, burden tests are more advantageous; by contrast, when effect-direction heterogeneity is present or when the proportion of non-causal variants is high, SKAT shows greater robustness through quadratic signal aggregation. Functional filtering and weighting optimization can improve the applicability of burden tests to some extent, whereas multi-kernel or adaptive methods (e.g., SKAT-O) can provide more robust performance when the underlying structure is unknown (Lee et al., 2014; Liu et al., 2019).
Therefore, variant aggregation design is not an independent step, but is tightly coupled with the choice of statistical method: aggregation strategy determines data structure, and data structure determines the optimal statistical test.
5 When to Choose Burden Tests or SKAT: A Genetic Architecture–Based Decision Framework
5.1 Structural basis for method selection
The choice between burden tests and SKAT essentially depends on the underlying genetic architecture within a variant set, particularly the causal proportion and effect-direction concordance. When biological prior knowledge supports the tendency of variants within the same gene or region to produce concordant effects—for example, protein-truncating variants in LoF-intolerant genes, or disruptive mutations within the same functional domain—and when functional annotation can effectively increase the proportion of causal variants, burden tests usually achieve higher statistical power. Under such circumstances, weighting strategies based on frequency and function (such as VT or joint weighting schemes) are often superior to fixed-threshold methods, and collapsing ultra-rare variants can help reduce estimation variance and improve stability (Lee et al., 2012a; Lee et al., 2012b).
By contrast, when effect directions within a set are uncertain or markedly heterogeneous—for example, in regulatory regions, cross-tissue regulatory effects, or mixed functional classes of variants—or when only a small number of sites are truly causal, the variance-component framework of SKAT is more advantageous. By accumulating squared effects, SKAT avoids cancellation among effects in opposite directions and maintains good robustness when the proportion of non-causal variants is high or when weighting schemes are misspecified (Lee et al., 2012a).
In practical studies, the true genetic architecture is often unknown or mixed. Therefore, SKAT-O, as an adaptive method, performs near-optimally in most scenarios. By making a data-driven trade-off between burden tests and SKAT, it achieves dynamic adaptation to different effect structures (Lee et al., 2012b). In settings where uncertainty in weights or kernel specification is high, multi-kernel strategies such as MK-SKAT can further alleviate performance fluctuations caused by model mismatch.
Sample structure also affects method choice. Under severe case-control imbalance or small sample size, SKAT-based methods are usually more stable than burden tests, but saddlepoint approximation or Firth correction is still needed to control the inflation of type I error (Zhang et al., 2019).
5.2 Decision expression for method selection
The above relationships can be uniformly understood as a two-dimensional decision problem driven by genetic architecture, with the core coordinates being effect-direction concordance × causal proportion. Within this space, different methods correspond to different optimal regions: when the causal proportion is high and effect directions are concordant, burden tests have a clear advantage; when causal signals are sparse or effect directions are heterogeneous, SKAT is more robust; and when the underlying structure is unknown or mixed, SKAT-O usually serves as the default strategy (Table 1).
.png) Table 1 Decision framework for method selection |
Therefore, method selection should not be regarded as a matter of tool preference, but rather as a statistical approximation to the underlying genetic model.
5.3 Methods in pactice: from set definition to robust inference
At the practical level, method selection is more appropriately organized as a layered decision pipeline rather than as a single-step model choice. This process can be summarized into three successive layers (Figure 2):
.png) Figure 2 Decision pipeline for method selection in rare variant association analysis (Methods in Practice) Note: This figure illustrates a decision pipeline for method selection in rare variant association analysis based on genetic architecture and data characteristics. The analysis begins with variant set definition (masking), including multiple MAF thresholds, functional annotation filtering, and ancestry-stratified frequency estimation. The core decision point is the evaluation of the genetic architecture within a gene region, particularly the causal proportion and effect-direction concordance. When the causal proportion is high and effect directions are concordant, burden tests usually have higher detection power; when effect directions are heterogeneous or causal variants are sparse, variance-component methods such as SKAT are more robust; when the structure is unknown or mixed, SKAT-O, as an adaptive method, is usually close to optimal. Furthermore, under conditions such as uncertainty in weight specification, multi-ancestry data, or complex sample structure (e.g., case–control imbalance or small sample size), inference robustness needs to be improved through strategies such as multi-kernel integration (MK-SKAT), saddlepoint approximation (SPA), or Firth correction. Finally, results should be reported in conjunction with multiple-testing correction, validation in independent cohorts, and interpretation based on functional annotation. This workflow emphasizes that method selection fundamentally depends on the underlying genetic architecture rather than on preference for a particular statistical tool |
(1) Variant set definition (masking)
The first step is to construct variant sets according to allele frequency and functional annotation, for example by using multiple MAF thresholds, functional-category filtering (e.g., LoF, deleterious missense, or regulatory variants), and ancestry-stratified frequency estimation. This step directly determines the causal proportion and signal-to-noise ratio and forms the basis for subsequent analyses.
(2) Genetic architecture assessment
Next, based on biological prior knowledge or preliminary statistical features, the effect structure within a set is evaluated. Scenarios with a high causal proportion and concordant effect directions are more suitable for burden tests; when the causal proportion is low or effect-direction heterogeneity is present, SKAT is preferred; when the structure is difficult to anticipate, SKAT-O provides a more robust compromise.
(3) Robustness adjustment
Under complex data conditions, further factors must be considered, including uncertainty in weight specification, sample imbalance, small-sample bias, and multi-ancestry differences. Correspondingly, corrections can be made through multi-kernel methods, saddlepoint approximation, or ancestry-stratified strategies in order to ensure the stability of statistical inference.
Overall, this process represents a stepwise constraint procedure from set construction → structure assessment → robustness control, with the goal of approximating the most suitable statistical model under unknown genetic mechanisms.
6 Simulation and Empirical Benchmarks: Structure-Driven Performance Evaluation
6.1 Simulation design
To systematically evaluate the performance of different methods under multiple genetic architectures, this study constructed a simulation framework covering causal proportion, effect-direction concordance, and the allele frequency spectrum. The core parameters included causal proportion (πc), directional concordance (θ), and the rare-variant spectrum (MAF spectrum), and different linkage disequilibrium (LD) structures and ancestry differences were introduced to simulate the complexity of real data. Phenotype types included quantitative traits and binary traits (including imbalanced case–control designs), and sample sizes were set to cover the typical ranges of both human and crop studies.
At the methodological level, burden tests (WSS and VT), SKAT, SKAT-O, and MK-SKAT were compared. Population structure was controlled within a mixed-model framework, and SPA or Firth correction was introduced for extremely imbalanced settings. Evaluation metrics included detection performance (power and type I error), statistical calibration (λGC and QQ plots), robustness (sensitivity to weights, LD, and ancestry), and computational cost.
6.2 Simulation results: structure-dependent performance
Simulation results showed that no method had a global advantage; rather, each method exhibited region-specific optimality under different genetic architectures.
When effect directions were concordant and the causal proportion was high, burden tests and VT achieved the lowest mean squared error. When effect directions were heterogeneous or signals were sparse, SKAT was more robust. Under unknown or mixed structures, SKAT-O was overall close to optimal. At the same time, burden tests were highly sensitive to weight and mask specification, whereas SKAT showed greater tolerance to model mismatch, and multi-kernel methods could further reduce fluctuations caused by uncertainty in model specification.
Different data systems also exhibited distinct patterns. In crop data with strong linkage disequilibrium (LD), coarse-grained aggregation tended to cause signal dilution and therefore required finer functional stratification. In human data, where ultra-rare variants are abundant, SKAT-based methods combined with robust correction were able to maintain good statistical properties. Multi-ancestry analyses, in turn, relied on stratified weighting and adaptive methods to maintain stability.
6.3 Empirical validation: from statistical signals to biological interpretation
In real data, the above patterns were further validated.
In analyses of crop disease resistance, coding-region variants, especially loss-of-function (LoF) variants, usually exhibited concordant effect directions, and burden tests were more likely to detect significant signals. In regulatory regions, by contrast, SKAT-based methods produced more stable results because effect directions were more complex. Human exome sequencing data likewise showed that burden tests performed particularly well in genes under strong functional constraint, whereas SKAT-O was more robust in variant sets containing multiple functional classes of variants.
In addition, under severe case–control imbalance, the introduction of SPA or Firth correction was crucial for controlling type I error. Taken together, these results indicate that method performance depends not only on effect structure, but is also significantly influenced by study design and data characteristics.
6.4 Summary
Combining simulation and empirical analyses, a consistent conclusion can be drawn: there is no universally optimal method in rare variant association analysis, and the optimal choice is determined by the underlying genetic architecture. Specifically, burden tests are suitable for scenarios with concordant effects and a high causal proportion, SKAT is suitable for structures with effect-direction heterogeneity or sparse signals, and SKAT-O provides a robust default choice when the underlying structure is unknown.
7 Discussion: A Unified Perspective from Aggregation Tests to Causal Structure Inference
7.1 The statistical essence of linear and quadratic aggregation
The difference between burden tests and the SKAT family does not fundamentally lie in the superiority or inferiority of particular statistical techniques, but rather in their different modeling assumptions about effect structure within a genomic region. The former, through linear aggregation of effects, essentially estimates the shift in the average effect within a variant set; the latter, by accumulating squared effects and treating variant effects as variance components of random variables, characterizes the dispersion of effect magnitude. The two statistics therefore correspond to different projections of “mean signals” and “variance signals.”
When effect directions are concordant and the causal proportion is high, linear aggregation can accumulate signals efficiently and thus achieve higher statistical power. By contrast, when effect directions are heterogeneous or causal variants are sparse, the quadratic form avoids effect cancellation and is therefore more robust. This division of roles indicates that performance differences between methods fundamentally arise from the degree to which they match the underlying genetic architecture, rather than from the superiority of one method over another (Lee et al., 2012a; Lee et al., 2014; Boutry et al., 2023a).
7.2 A structural interpretation of method complementarity
Based on the above understanding, the “complementarity” between burden tests and SKAT can be reinterpreted as the optimal approximation of different statistical models across different regions of genetic architecture. In the decision pipeline described above, this relationship is formalized as a structural space jointly determined by causal proportion and effect-direction concordance. Within this space, burden tests correspond to scenarios in which the structure is known and concordant effects dominate, SKAT provides robust estimation for heterogeneous structures, and SKAT-O achieves adaptive approximation to unknown structures through a data-driven balance between the two.
Therefore, method selection should no longer be regarded as an empirical problem of tool comparison, but rather as a problem of statistical identification of the underlying causal structure. In other words, the core task of rare variant association analysis is to reasonably approximate the effect structure within a variant set under conditions of limited sample size and incomplete information. This perspective elevates the traditional problem of “method comparison” into a unified framework of structure-aware inference.
7.3 Improving robustness: from adaptive methods to model integration
In real data, genetic architecture is often mixed or unknown, and robustness therefore becomes a key consideration in method selection. By adaptively weighting burden tests and SKAT, SKAT-O achieves near-optimal performance across a wide range of scenarios and can therefore serve as a default analytical strategy (Lee et al., 2012b; Moutsianas et al., 2015). However, when there is substantial uncertainty in weight specification or kernel choice, a single model may still be biased.
In this context, multi-kernel methods such as MK-SKAT reduce the risk caused by model mismatch by integrating different weighting schemes and kernel functions; in essence, this represents a shift from single-model selection to model integration. By combining the advantages of these approaches, it is possible to formulate a practical strategy that balances detection power and interpretability: using SKAT-O for initial screening, applying multi-kernel methods for robust estimation in candidate regions, and then using burden tests to verify the presence of concordant effects. This workflow achieves a relative balance between statistical efficiency and biological interpretability.
7.4 Cross-system consistency: a unified mechanism in human and crop genetics
Although these methods were originally developed in human genetics, their applicability shows a highly consistent dependence on genetic structure across different biological systems. In crop systems, stronger linkage disequilibrium and allele clustering effects often cause signals to spread across larger genomic intervals, thereby aggravating signal dilution in burden tests. However, in functionally well-defined coding regions, such as key resistance genes or metabolic pathway genes, concordant effects still give burden tests an advantage. By contrast, regulatory regions are more suitable for SKAT-based methods because of the complexity of effect directions and underlying mechanisms.
In human data, rare loss-of-function variants often correspond directly to clear biological mechanisms, and burden tests therefore perform particularly well in such regions. In contrast, for variant sets containing multiple functional categories or complex regulatory effects, SKAT and its extensions provide more robust statistical inference. Overall, differences across systems arise mainly from differences in genetic architecture and variant distribution, rather than from limitations in the applicability of the methods themselves.
7.5 Key challenges and future directions
Although existing methods have shown good performance across a variety of scenarios, several structural issues still limit their further development. First, uncertainty remains in variant annotation and set-construction strategies, especially in noncoding regions and tissue-specific regulation, which directly affects the estimation of causal proportion and statistical power. Second, substantial differences in allele frequencies and linkage disequilibrium structures across ancestries may reduce model stability and increase the risk of false positives.
At the statistical level, under conditions involving extremely low-frequency variants and sample imbalance, traditional asymptotic approximations often fail, making methods such as saddlepoint approximation or Firth correction necessary to maintain inferential accuracy (Zhang et al., 2019). In addition, there is currently a lack of unified analytical standards, including definitions of variant sets, weighting choices, and result-reporting criteria, which to some extent limits the reproducibility and comparability of results across studies.
Future research needs to advance in three directions: first, improving functional annotation and set-construction strategies in order to enrich causal variants; second, developing robust methods suitable for multi-ancestry data and complex sample structures; and third, promoting the standardization of analytical workflows and reporting criteria so as to improve the overall reproducibility and interpretability of rare variant studies.
8 Conclusion
From a unified statistical genetics perspective, this study systematically clarifies the intrinsic relationship between burden tests and the SKAT family in rare variant association analysis. The results show that these two classes of methods are not interchangeable alternatives, but rather correspond to different assumptions about causal structure: burden tests characterize genetic patterns dominated by concordant effects through linear aggregation, whereas SKAT accommodates heterogeneous effect directions and sparse causal scenarios through variance-component modeling. Therefore, differences in method performance fundamentally arise from genetic architecture rather than from statistical techniques themselves.
On this basis, this study further proposes that method selection should shift from an empirically driven process to a structure-driven one. By incorporating factors such as variant set definition, effect-direction concordance, and causal proportion into a unified decision framework, the question of “which method should be chosen” can be transformed into “which causal structure should be identified.” In real-world settings where the underlying structure is unknown or mixed, SKAT-O, as an adaptive method, can achieve near-optimal performance across different structural patterns, while multi-kernel integration provides a more robust extension for dealing with uncertainty in weighting schemes and model specification.
Both simulation and empirical results show that there is no single method that is superior in all scenarios; instead, there exist “optimal regions” defined by genetic architecture. This finding is consistent with the layered decision pipeline proposed in this study and supports the adoption of a strategy of “screening-refinement-validation” in practical research in order to balance detection power, robustness, and interpretability.
From a broader perspective, rare variant analysis adds a critical layer to complex trait genetics. Compared with single-variant association analysis and probability-based fine-mapping, its central role lies in characterizing effect structure at the gene-region level, thereby statistically connecting variant sets with functional mechanisms. The introduction of this layer enables research on complex traits to move further from isolated association signals toward structured causal inference.
Overall, the rare variant analysis framework centered on aggregation tests provides a structure-oriented inferential tool for complex trait genetics. The key task for future research is to further improve the ability to identify complex effect structures in the context of accumulating multi-ancestry and multi-omics data, while maintaining model robustness and the interpretability of results.
Author Contributions
Xuanjun Fang conducted this study, including literature review, data analysis, and the writing and revision of the manuscript. The author has read and approved the final version of the manuscript.
Acknowledgements
This work was supported by a Major Project of the National Natural Science Foundation of China (No. 30490254).
Bigdeli T.B., Neale B.M., and Neale M.C., 2014, Statistical properties of single-marker tests for rare variants, Twin Research and Human Genetics, 17(3): 143-150.
https://doi.org/10.1017/thg.2014.17
Boutry S., Helaers R., Lenaerts T., and Vikkula M., 2023a, Rare variant association on unrelated individuals in case-control studies using aggregation tests: existing methods and current limitations, Briefings in Bioinformatics, 24(6): bbad412.
https://doi.org/10.1093/bib/bbad412
Boutry S., Helaers R., Lenaerts T., and Vikkula M., 2023b, Excalibur: A new ensemble method based on an optimal combination of aggregation tests for rare-variant association testing for sequencing data, PLoS Computational Biology, 19(9): e1011488.
https://doi.org/10.1371/journal.pcbi.1011488
Chen H., Lumley T., Brody J., Heard-Costa N.L., Fox C.S., Cupples L.A., and Dupuis J., 2014, Sequence kernel association test for survival traits, Genetic Epidemiology, 38(3): 191-197.
https://doi.org/10.1002/gepi.21791
Chen H., Meigs J.B., and Dupuis J., 2013, Sequence kernel association test for quantitative traits in family samples, Genetic Epidemiology, 37(2): 196-204.
https://doi.org/10.1002/gepi.21703
Falk I., Zhao M., Nait Saada J., and Guo Q., 2023, Learning the kernel for rare variant genetic association test, Frontiers in Genetics, 14: 1245238.
https://doi.org/10.3389/fgene.2023.1245238
Fang X.J., and Wu W.R., 2026, Evolution of statistical genetic paradigms: from linkage analysis and candidate gene strategies to GWAS, Fenzi Zhiwu Yuzhong (Molecular Plant Breeding), 24(9): 2817-2829.
Guo M.H., Plummer L., Chan Y.M., Hirschhorn J.N., and Lippincott M.F., 2018, Burden testing of rare variants identified through exome sequencing via publicly available control data, The American Journal of Human Genetics, 103(4): 522-534.
https://doi.org/10.1016/j.ajhg.2018.08.016
Hecker J., Townes F.W., Kachroo P., Laurie C., Lasky-Su J., Ziniti J., Cho M., Weiss S., Laird N., and Lange C., 2020, A unifying framework for rare variant association testing in family-based designs, including higher criticism approaches, SKATs, and burden tests, Bioinformatics, 36(22-23): 5432-5438.
https://doi.org/10.1093/bioinformatics/btaa1055
Ionita-Laza I., Lee S., Makarov V., Buxbaum J.D., and Lin X., 2013, Sequence kernel association tests for the combined effect of rare and common variants, The American Journal of Human Genetics, 92(6): 841-853.
https://doi.org/10.1016/j.ajhg.2013.04.015
Jiang Z., Zhang H., Ahearn T.U., Garcia-Closas M., Chatterjee N., Zhu H., Zhan X., and Zhao N., 2023, The sequence kernel association test for multicategorical outcomes, Genetic Epidemiology, 47(6): 432-449.
https://doi.org/10.1002/gepi.22527
Lee S., Abecasis G.R., Boehnke M., and Lin X., 2014, Rare-variant association analysis: study designs and statistical tests, The American Journal of Human Genetics, 95(1): 5-23.
https://doi.org/10.1016/j.ajhg.2014.06.009
Lee S., Emond M.J., Bamshad M.J., Barnes K.C., Rieder M.J., Nickerson D.A., Christiani D., Wurfel M., and Lin X., 2012b, Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies, The American Journal of Human Genetics, 91(2): 224-237.
https://doi.org/10.1016/j.ajhg.2012.06.007
Lee S., Wu M.C., and Lin X., 2012a, Optimal tests for rare variant effects in sequencing association studies, Biostatistics, 13(4): 762-775.
https://doi.org/10.1093/biostatistics/kxs014
Liu R., Yuan M., Xu H., Chen P., Xu X.S., and Yang Y., 2020, Adaptive weighted sum tests via LASSO method in multi-locus family-based association analysis, Computational Biology and Chemistry, 88: 107320.
https://doi.org/10.1016/j.compbiolchem.2020.107320
Liu Y., Chen S., Li Z., Morrison A.C., Boerwinkle E., and Lin X., 2019, ACAT: a fast and powerful p value combination method for rare-variant analysis in sequencing studies, The American Journal of Human Genetics, 104(3): 410-421.
https://doi.org/10.1016/j.ajhg.2019.01.002
Moutsianas L., Agarwala V., Fuchsberger C., Flannick J., Rivas M.A., Gaulton K.J., Albers P., McVean G., Boehnke M., Altshuler D., and McCarthy M.I., 2015, The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease, PLoS Genetics, 11(4): e1005165.
https://doi.org/10.1371/journal.pgen.1005165
Oualkacha K., Dastani Z., Li R., Cingolani P.E., Spector T.D., Hammond C.J., Richards J., Ciampi A., and Greenwood C.M., 2013, Adjusted sequence kernel association test for rare variants controlling for cryptic and family relatedness, Genetic Epidemiology, 37(4): 366-376.
https://doi.org/10.1002/gepi.21725
Pan W., Kim J., Zhang Y., Shen X., and Wei P., 2014, A powerful and adaptive association test for rare variants, Genetics, 197(4): 1081-1095.
https://doi.org/10.1534/genetics.114.165035
Rajabli F., and Kunkle B.W., 2023, Strategies in aggregation tests for rare variants, Current Protocols, 3(11): e931.
https://doi.org/10.1002/cpz1.931
Wang X., 2014, Firth logistic regression for rare variant association tests, Frontiers in Genetics, 5: 187.
https://doi.org/10.3389/fgene.2014.00187
Wu B., and Pankow J.S., 2016, Sequence kernel association test of multiple continuous phenotypes, Genetic Epidemiology, 40(2): 91-100.
https://doi.org/10.1002/gepi.21945
Zhang X., Basile A.O., Pendergrass S.A., and Ritchie M.D., 2019, Real world scenarios in rare variant association analysis: the impact of imbalance and sample size on the power in silico, BMC Bioinformatics, 20(1): 46.
https://doi.org/10.1186/s12859-018-2591-6
Ziyatdinov A., Mbatchou J., Marcketta A., Backman J., Gaynor S., Zou Y., Joseph T., Geraghty B., Herman J., Watanabe K., Ghosh A., Kosmicki J., Locke A., Thornton T., Kang H., Ferreira M., Baras A., Abecasis G., and Marchini J., 2024, Joint testing of rare variant burden scores using non-negative least squares, The American Journal of Human Genetics, 111(10): 2139-2149.
https://doi.org/10.1016/j.ajhg.2024.08.021




. HTML
Associated material
. Readers' comments
Other articles by authors
. Xuanjun Fang
Related articles
. Rare variants
. Burden test
. SKAT
. SKAT-O
. Aggregation tests
. Causal architecture
. Decision pipeline
. Complex trait genetics
Tools
. Post a comment